Cell heterogeneity is essential for the physiological functions of complex tissues under normal and disease conditions. Systematic interpretation of heterogeneous cell populations and precise identification of key cell subtypes hold substantial importance in gaining critical insights into tissue development, physiological functions, cellular interactions, and the dynamic progression of diseases. Transcriptome profiling at single-cell resolution has been a common practice for dissection of cell heterogeneity. However, almost all of these previous studies have solely relied on the overall expression levels of genes to assess cell heterogeneity in complex tissues. The canonical approach completely ignores the RNA splicing patterns, due to a series of methodological challenges.
Variations in RNA splicing serve as a major source of the extensive transcriptome complexity in mammalian species. Via different splicing patterns, a single gene can generate multiple distinct transcript variants, leading to a remarkable expansion of complexity in both the transcriptome and proteome. This process underscores the diversity and plasticity exhibited by gene expression and function. Consequently, the comprehensive analysis of cellular heterogeneity in complex tissues necessitates the incorporation of RNA splicing profiles.
In response to this pressing demand, the research team led by Xuerui Yang at the School of Life Sciences, Tsinghua University, have developed a bioinformatics method SCASL (single-cell clustering based on alternative splicing landscapes) for classifications of cell identities by interrogating the RNA splicing heterogeneity in single cells. SCASL facilitates a comprehensive and quantitative analysis of single-cell RNA splicing heterogeneity, providing a new scheme for classifications of cell identities based on AS profiles. The team's pioneering research has been published in Nature Communications on March 9th, under the title "Interrogations of single-cell RNA splicing landscapes with SCASL define new cell identities with physiological relevance."
SCASL relies on de novo identifications of alternative splicing (AS) events from single-cell RNA-seq data, which is annotation-free and allows comprehensive survey of all AS schemes. SCASL effectively resolves the issue of biased and sparse data coverage on scRNA splicing by employing iterative kNN imputation. By employing spectral clustering, SCASL achieves unsupervised classifications of cell identities and generates a comprehensive landscape depicting the heterogeneity of scRNA splicing. Importantly, when applied to the same scRNA-seq data, most of the clustering results from SCASL are not readily available via conventional cell clustering based on single-cell gene expression data.

Figure 1. Schematic overview of the SCASL pipeline
Via a series of novel strategies for data imputation and clustering, SCASL recovered new classifications of cell subpopulations with high physiological relevance, under contexts of tumorigenesis, tissue development, and tumor microenvironment. For example, tumor cells are known for their intrinsic global gene expression disorder and stochasticity. Previous studies based on single-cell gene expression profiles have encountered major challenges for classifications of intra-tumor cell subtypes with physiological functions. Based on RNA splicing landscapes, SCASL clearly identified the cell populations at intermediate stages of tumorigenesis, which have not been reported before, for example precancerous normal epithelial cells, early-stage tumor cells, and cells of micrometastasis in TNBC.
In the context of normal tissue development, SCASL also demonstrates unique advantages. Specifically, SCASL successfully illustrates two cell lineages of cholangiocytes and hepatocytes during embryonic liver development, enabling precise identification of transitional clusters within the lineage. More interestingly, the single-cell RNA splicing landscape revealed a special cluster, which was composed of annotated cholangiocytes but positioned between the two major developmental lineages, exhibiting functional characteristics of both cell types simultaneously. This intriguing finding suggests that this subtype of cells may serve as a potential crosstalk bridging the transition between the two cell lineages during liver development.
Link to the article: https://www.nature.com/articles/s41467-024-46480-9
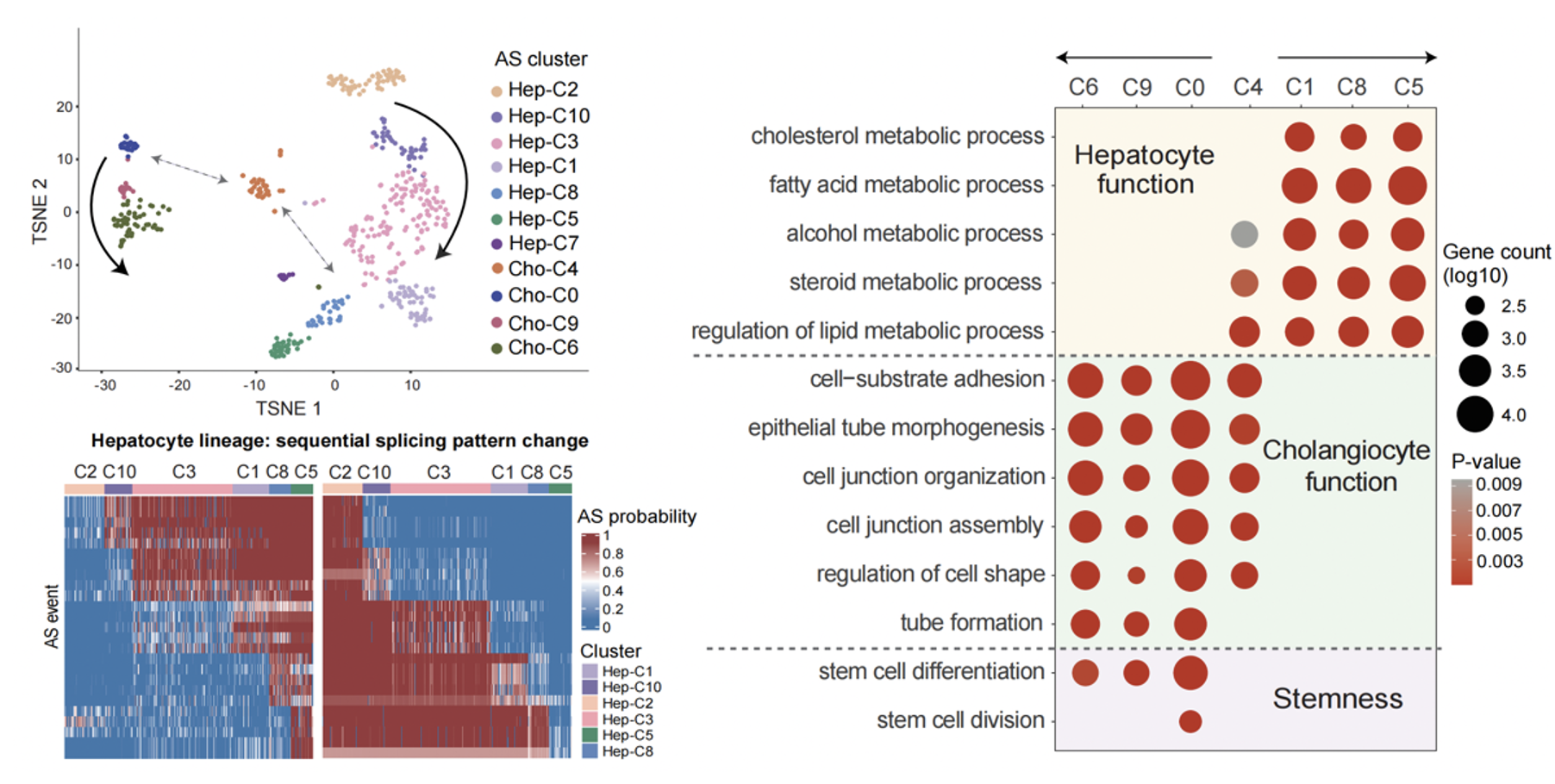
Figure 2. Reconstruction of mouse embryonic liver developmental lineages by SCASL (left); C4 illustrates the potential crosstalk between two cell lineages (right).
In summary, the intrinsic RNA splicing heterogeneity provides critical information for precise definitions of physiologically relevant cell subtypes. Xuerui Yang’s work presents a new method SCASL for mining such information. New insights from this pipeline could greatly help understanding the functions and progressive developments of complex tissues.
The paper's first author is Xianke Xiang, a Ph.D. student from the School of Life Sciences at Tsinghua University. Xuerui Yang, an Associate Professor, serves as the corresponding author. Dr. Zemin Zhang, a professor at the Biomedical Pioneering Innovation Center, Peking University, and his team made significant contributions by providing essential data and tools for this research. This work was funded by the National key research and development program, the National Natural Science Foundation of China, and the Tsinghua University Initiative Scientific Research Program. Moreover, the research received support from the Tsinghua University Branch of China National Center for Protein Sciences (Beijing) and Tsinghua University Technology Center for Protein Research, including the core facilities of Biocomputing, Genome Sequencing and Analysis at Tsinghua University.